


设计和开发
在设计和开发产品时应考虑UDI系统的目标和预期效果。在将器械投放市场或向公告机构提交技术文件进行合格评估之前,应使用发行实体的规则确保UDI-DIs的分配。此外,UDI-PI应正确复制生产标识符,根据风险管理或法规要求分配到设备标签上。可追溯性、产品序列化也应在适当的风险管理、法规要求(如活性植入物需要序列号)和其他利益相关方/国家监管机构(如注册中心)的期望或要求的基础上进行。
用于分配UDI、UDI的变更和相应的可追溯程度的程序应该被适当地记录下来。
产品文档和保留
作为技术文档的一部分,应保持其指定的所有UDI (UDI-DIs和UDI-PIs) 的最新列表。应在最后一个欧盟符合性声明涵盖的设备进入欧盟市场后至少10年向药监当局提供技术文件。在可植入设备的情况下,期限应在最后一个设备被投放在欧盟市场之后至少15年。
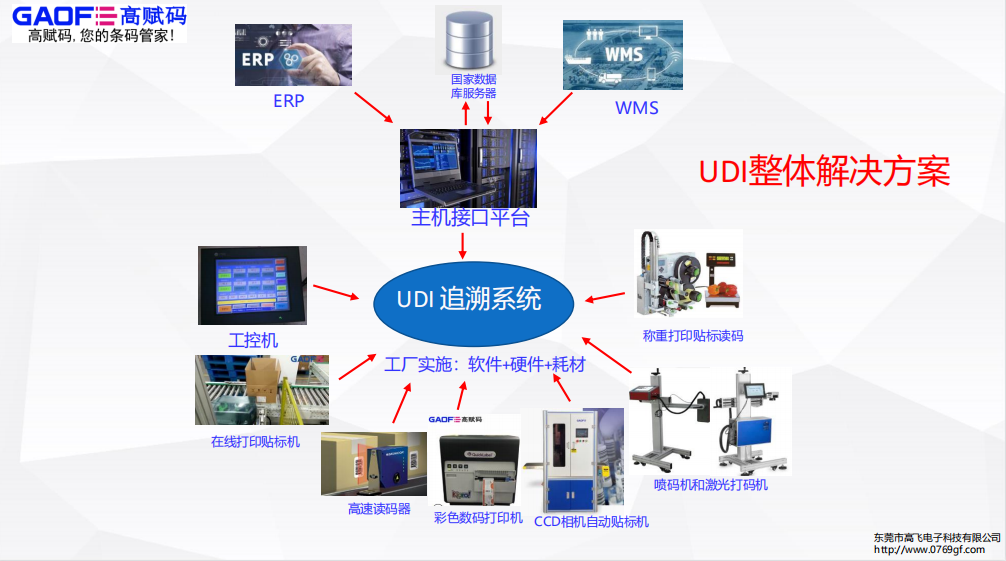
生产和过程
可以考虑从生产阶段开始使用UDI,以确保有效的产品信息管理。例如,UDI-DIs可以用作参考或目录编号。
应根据MDR/IVDR的要求,针对每个单独的类型/型号,根据每个风险级别的不同时间表,决定何时、何地和如何应用UDI载体。
由于UDI载体可能会对制造过程产生影响,对于直接标记的器械,应提前确定是否适用规定的任何豁免,以确保在设备投放到欧盟市场时满足要求。例如,在MDR/IVDR下,如果不能在某些设备上进行直接标记,则对直接标记有豁免。这种豁免应记录在案,最好是在技术文件中。此外,设备包级别将需要有UDI载体。
标签注意事项:
确保标签印刷过程经过验证,器械的使用和维护符合相关程序。
评估已验证工艺的变更对器械标签的影响。
用于实现UDI系统的软件(如UDI贴标设备和包装,机器到机器自动上传UDI数据到EUDAMED)应按照相关程序保持有效状态。
严重事件和现场安全纠正措施
UDI应被用于报告严重事件和现场安全纠正措施。组织的内部程序应详细说明这些要求。
采购控制
虽然采购部件不受UDI义务约束,除非它们按照MDR和IVDR作为医疗设备进行监管,但应基于以下考虑采购程序:
调查现有的采购活动及其控制,确定可能影响UDI系统符合性的材料和设备。包括标签材料、打印机和扫描仪。
调查采购活动,确定哪些部件、材料和设备是作为器械投放市场的,并承担制造商应尽的 义务。
对受UDI义务约束的货物的采购进行识别和过程/记录。
文件和记录
器械的Basic UDI-DI应出现在:
警戒和上市后监测报告(如MIR和PSUR),
欧盟符合性声明,
技术文件,
安全和临床性能总结,
自由销售证书,
部分EC证书,如EU技术文件评估证书,EU型式检查证书和EU产品验证证书。UDI应包括在植入卡上。内部程序应参考上述案例中基本UDI的出现。
器械的UDI(UDI-DI+UDI-PI)应出现在:
出现在标签上或设备本身(如适用),以及所有更高级别的包装上;
参考技术文档。
而Basic UDI-DI和UDI-DI应以EUDAMED格式提供给UDI数据库,只有在出现警戒问题时,才应将UDI-PI提供给EUDAMED。
企业资源计划ERP
如使用ERP系统来捕获UDI数据,应保持并确认用于连接操作打印机的软件,收集UDI元数据并创建UDI,验证M2M连接到EUDAMED和其他步骤的文档。
提供给EUDAMED数据库的UDI数据
根据MDR和IVDR,在一个设备投放欧盟市场之前,除了定制的、调查的或性能研究的设备,应确保与相关设备相关的信息正确提交并转移到EUDAMED的UDI数据库中。制造商应更新提交给EUDAMED的信息。
综上所述,UDI系统不单是在医疗设备本身、标签、包装上印刷一维或二维码,UDI系统影响着质量管理体系中的各部分,所以需要各医疗器械厂商将UDI系统整合到质量体系中。
